Chapitre 3 : cartographie du génome humain
Le but dans ce chapitre est d’établir une carte très précise des différents gènes sur le trois milliards de paires de bases du génome humain. Pour cela, 4 approches complémentaires sont nécessaires :
- cytogénétique : qui permet de localiser les gènes sur des bandes ou des sous bandes du chromosome (on localise un gène chr11q1.3.2, soit bras long du chromosome 11, région 1 à partir du centromère, bande 3 et sous bande 2).
- Génétique : on établit une carte des lésions génétiques où les distances sont exprimées en fréquence de recombinaison par crossing-over. Cela permet une estimation d’une distance entre les gènes.
- Moléculaire : à partir d’une banque d’ADNg, on va séquencer les clones de la banque et on va recoller les séquences (par informatique) dans leur ordre et leur sens.
- Bioinformatique : dans ce magma de trios milliards de pares de bases, des programmes vont chercher les 50 000 gènes humain
- Préparation des chromosomes métaphasique ou des noyaux interphasiques
- Dénaturation de l’ADNg chimiquement ou physiquement
- Hybridation quelques heurs à trois jours avec une sonde fluorescente.
- Lavage puis révélation
- centromérique (séquence CEN)
- subtelomérique (séquence TEL)
- locus spécifique sonde spécifique d’un gène.
- Spécifique des bandes d’un seul chromosome (plusieurs sondes, toutes marquées par un fluorochrome différent)
- Multiplex FISH (plusieurs sondes marquées avec des fluorochromes différents et se fixant chacune sur un seul chromosome)
Sonde PCR-alu : ce sont des sondes fabriquées par PCR en utilisant des amorces reconnaissant 2 séquences successives.
Les sondes peuvent aussi être fabriquées par Nick translation, par random priming sur les clones d’ADNg (cosmides, PAC, BAC, YAC)
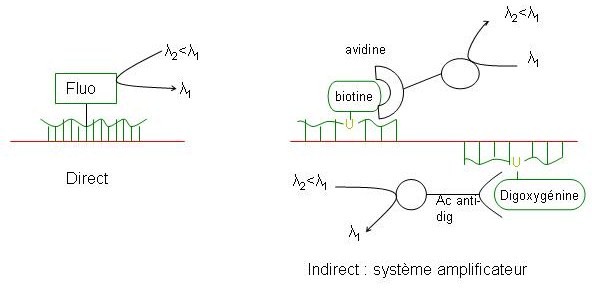
Révélation des sondes
Le principe consiste à cohybrider un ADN normal de référence marqué par un fluorochrome 1 et l’ADN du patient marqué par un fluorochrome 2. Ces 2 ADN marqués différemment sont mis en contact soit avec des chromosomes normaux de références (CGH conventionnelle), soit avec des clones BAC ou PAC d’une banque d’ADNg (CGH array)
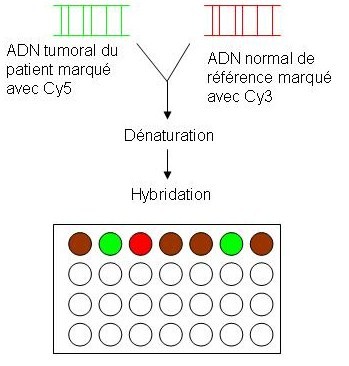
Légende : marron = séquence ADN normal. Dominance de vert = excès d’hybridation de l’ADN tumoral (répétition). Dominance de rouge : défaut d’hybridation de l’ADN tumoral (délétion)
Les spots sont lus par un scanner laser, qui transmet les résultats pour traitement informatique. Les spots de couleurs verts et rouges indiquent au manipulateur qu’il y a une différence d’hybridation entre l’ADN normal et l’ADN malade. Comme l’ordre et l’identité des BAC, YAC ou PAC est connu, on peut remonter à la séquence défectueuse chez le malade.
On considère que la probabilité de recombinaison par crossing-over pendant la méiose entre deux locus génétique dépend de la distance physique qui les sépare. Si deux locus sont éloignés, la probabilité de crossing-over augmente. On dit qu’il y a absence de liaison entre les deux gènes. Si deux locus sont rapprochés, la probabilité de crossing-over diminue. On dit que les gènes sont liés.
On va faire de multiples analyses familiales et étudier la fréquence de recombinaison entre deux locus.
- le premier locus est le gène d’intérêt à localiser (en général un gène morbide)
- le second locus est un marqueur génétique, qui est spécifique à chaque individu et facilement analysable.
Le but de cette approche est d’établir une carte de liaisons génétiques où les distances sont exprimées en centimorgan (CM) qui est l’unité de recombinaison.
Définition : il s’agit des SNP (single nucleotide polymorphism), c'est-à-dire de variations ponctuelles de séquences touchant un seul nucléotide au niveau du locus donné.
Pour analyser facilement les SNP, ont choisi ceux qui sont à l’intérieur d’un site de restriction. On les appelle alors les RFLP (restriction fragment lenght polymorphism), soit polymorphisme de longueur de fragments de restriction.
1er méthode : on fait une extraction d’ADNg, suivit d’un clivage par une enzyme de restriction qui touche le RFLP, puis une électrophorèse (obtention d’un SMIR), transfert sur membrane, puis hybridation avec une sonde à cheval sur le RFLP (figure2-3)
2nd méthode : on amplifie par PCR le RFLP en utilisant des amorces de part et d’autre du RFLP et ensuite on live les amplicons obtenus par l’enzyme de restriction touchant le RFLP.
Les RFLP ont une faible informativité (le PIC=0,38) car ils sont seulement biallélique. Néanmoins, ils permettent de se repérer sur le chromosome (sorte d’étiquettes) et d’estimer la distance entre un gène morbide et un RFLP.
L’allèle dominant D de la maladie est lié à la forme clivé du RFLP, alors que l’allèle récessif d de la maladie est lié à l’allèle non clivé du RFLP. Le seul crossing-over qui c’est produit dans cette famille touche l’enfant 8 et permet le calcul de la faible distance entre le gène d et le RFLP.
Il s’agit d’un motif nucléotidique de base répété en tandem un nombre de fois variable. Il s’agit des VNTR et STR. Ils sont très informatif car polyallélique (PIC environ de 1). (figure5)
On fait de multiples analyses familiales en calculant la fréquence de recombinaison entre le locus morbide et un marqueur génétique (RFLP, STR, VNTR…)
Et L(θ0,5) : vraisemblance que les deux locus soient indépendants.
On trace ensuite la courbe Z(θ)=f(θ) et on a trois cas de figure. (figure6-7)
- Zθ>3, la vraisemblance gène lié est au moins mille fois supérieur à la vraisemblance gène non lié = les deux gènes sont liés, donc très proche, et leur distance correspond au θ du Z(θ)max
- Zθ<-2, les gènes sont indépendants, donc très éloignés
- Zθ=0,5. Zθ n’est pas supérieur à trois ni inférieur à -2 = ininterprétable, il faudra conduire de nouvelles études.
Les distances sont exprimées en Morgan, ou centimorgan (CM), c’est l’unité de recombinaison qui correspond à l’intervalle où se produit obligatoirement un crossing-over par méiose.
On a environ 60 chiasmas (cicatrices de crossing-over)/méiose sur un ADN diploïde
Soit 60 morgan/ ADN diploïde
30 morgan/ ADN haploïde de 3.109 pb
1 morgan/ 108 pb
1CM/106 pb
On aboutit aux cartes de liaisons génétiques où les gènes sont localisés à 1CM près, soit à 106 pb près (figure8).
Le but ici sera d’établir la séquence exacte du génome au nucléotide près.
- la longueur du génome humain : on ne peut pas séquencer un chromosome en entier, d’un seul coup, car une réaction de séquençage permet de lire au maximum 600pb. Le nombre de séquençage à réaliser est énorme, et il faudra « recoller » intellectuellement les séquences dans l’ordre = l’homme fait donc appelle à des automates (séquençage), puis à l’informatique.
- Les polymérases utilisées par le séquençage font malheureusement des erreurs. Pour être sur d’avoir la bonne séquence, il faut séquencer environ dix fois la même région (soit environ 50 millions de réactions de séquençage)
- L’ADN est rempli de séquences répétitives qui vont provoquer des alignements erronés entre des séquences issues de chromosomes différents.
- la mise en ordre des clones, puis une fois trié, ces derniers sont séquencés
Dans les deux cas, la technique de séquençage utilisé est du Sanger fluorescent.
Dans cette réaction, on interrompt l’élongation par des didesoxyribonucléotides (ddNT). Pour éviter une révélation radioactive des fragments interrompus, on utilise deux techniques fluorescentes.
Les amorces sont fluorescentes « dye primer »
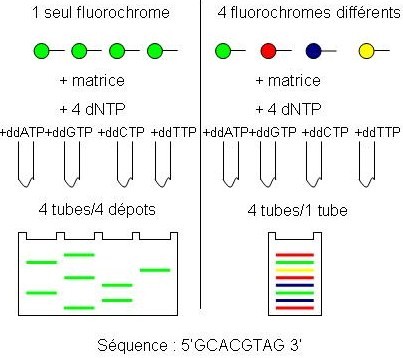
Les ddNTPs sont fluorescents (dye terminator)
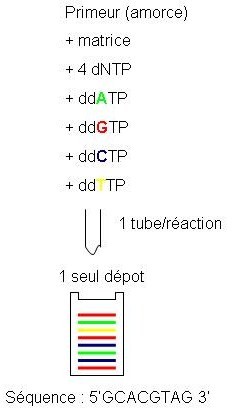
Une diode laser balaye une fenêtre en bas du gel et excite avec une seul ou quatre longueur d’onde différente(s) les fluorochromes. Les longueurs d’ondes d’émission passent par un filtre et un photomultiplicateur qui envoie l’information à un ordinateur, visualisant à l’écran chaque lettre par un pic de couleur.
On séquence tout les clones de la banques puis on ordonne ces séquences en s’aidant des régions chevauchantes entre les clones. On obtient un ensemble de clones « recollé » ensemble appelé contigs.
Parfois on a du mal a recoller dans l’ordre les contigs à cause de l’ADN répétitif. On utilise alors des clones possédant des lectures d’extrémités appariées. Il s’agit de clones ayant, à une extrémité de l’insert, 600 pb appartenant à un premier contig (A) et ayant à l’autre extrémité de l’insert 600 pb appartenant à l’autre contig (B). Ceci permet de recoller les contigs A et B et d’obtenir une séquence plus longue appelée montage.
Soit plusieurs séquences donnent un contig, et plusieurs contigs donnent un montage.
La stratégie est l’inverse de la précédente. On ordonne les clones avant de les séquencer. Pour les ordonner, on utilise des empreintes de restriction.
On compare les fragments de restriction obtenue sur chaque clone de la banque. On estime que deux clones sont chevauchants lorsqu’ils ont au moins 50% de fragments de restriction en commun. Ceci permet d’obtenir une succession de clones ordonnés et orientés.
C’est ce que l’on appelle un contig de clones ou carte physique du génome. On choisit ensuite des clones ayant un chevauchement minimum pour limiter au maximum le nombre de réaction de séquençage à faire. L’ensemble de ces clones choisit constitue le tuilage minimum. Les clones sont clivés en sous clones, qui sont séquencés puis récolés entre eux.
Cette approche génétique nous permet donc de connaître la séquence au nucléotide près. Reste à trouver la position des gènes dans ce magma de lettre A, T, C et G.
Il existe différents programmes permettant de localiser les 50 000 gènes humains.
L’ordinateur recherche toute séquence commençant par un codon ATG (codon initiation) et finissant par un des trois codons stop, avec une séquence multiple de trois nucléotides entre les deux. Cette recherche ce fait dans les six cadres de lectures (3 cadres par brin)
Si un alignement est possible entre un ADNc full-lenght et des régions successives sur l’ADNg, alors l’ORF candidate devient une ORF sur. Il existe même des banques informatiques contenant uniquement des séquences 3’ et 5’ des ADNc : EST (Expressed Sequence Tag). Les EST permettent de localiser le début et la fin d’un gène sur l’ADNg avec le bon cadre de lecture.
On demande à l’ordinateur de recherché les sites de fixation des sites de régulateurs dans le promoteur, les signaux d’épissage, le site de polyadénilation dans toute les ORF candidates. Seuls les ORFs ayant des signaux sont retenues.
Les ORF candidates sont introduites dans des bases publiques de données, soit sous forme de séquence nucléotidique (BLASTn), soit sous forme de séquence AA (BLASTp). Le programme recherche les alignements possibles entre la séquence candidate et les séquences contenues dans la base.
- deux gènes ou protéines se ressemblant et de la même espèce : paralogue
- eux gènes ou protéines se ressemblant et d’espèce différente : orthologue
Dans chaque séquence, les codons synonymes ne sont pas utilisés de manière équilibrée.
Ex : drosophile : codon UGC (cyst) : 73%, et codon UGU (cyst) : 27%.
Si dans une ORF candidate, les codons sont utilisés comme le prévoit l’espèce, l’ORF proposée à de forte chance d’être authentique.